AI-driven drug discovery with transformer models
The “Lock and Key” Challenge
Drug discovery is costly and time-consuming; worldwide pharmaceutical R&D spending reached an estimated USD 190 billion in 2024, with development times for new drugs typically ranging from 10 to 15 years.
Developing drugs that interact with a specific target involved in a disease, such as an enzyme or receptor, is a process of designing, screening, and refining molecules to bind to that target. We can imagine the challenge for researchers as being like designing a key for a lock, but where both the key and the lock are constantly wiggling and changing shape. Furthermore, the key may often have to hold two or more locks together simultaneously. This is known as a ternary complex.
Take the example of Rapamycin, a small molecule drug originally discovered in a soil sample from Easter Island, produced by the bacterium Streptomyces hygroscopicus. It is used as an immunosuppressant to prevent organ transplant rejection and as a treatment for cancers such as renal cell carcinoma. Intriguingly, Rapamycin is also known to extend lifespans in mice and has generated great interest for its potential to extend human longevity. Rapamycin acts as a molecular glue between a large kinase protein called the mechanistic Target of Rapamycin (mTOR), often referred to as the cell’s “growth switch”, and FKBP12, which is involved in the human immune response.
Beyond AlphaFold: Open-Source Drug Discovery with Boltz-2
Running an experiment to determine the interaction between a drug candidate and a target protein, or a ternary protein complex, can take weeks or months and cost thousands of dollars. With the latest advances in transformer-based foundational models like AlphaFold 3 and Boltz-2, these interactions can be predicted in seconds, with thousands of drug candidates screened in parallel using this in silico approach.
While the license for AlphaFold 3 is restrictive for commercial use, Boltz-2 is open source and licensed under the highly permissive MIT license. This allows clients to run their drug pipelines on their own private infrastructure, protecting valuable intellectual property. This ensures data sovereignty, guaranteeing that proprietary lead compounds and intellectual property never leave your private infrastructure.
Boltz-2 uses a deep learning approach called co-folding to simulate how multiple proteins interact with a drug candidate. It is particularly useful for:
- Hit discovery: Rapidly triaging large chemical libraries to effectively distinguish potential ‘binders’ from ‘decoys’ during high-throughput screening.
- Lead optimization: The affinity head allows researchers to rank close analogs and predict how small molecular modifications will impact potency.
- Difficult pockets: Simulating the flexible shapes of ligands and targets simultaneously.
The Boltz-2 architecture integrates many recent advances in machine learning, such as a diffusion/denoising module, similar to Midjourney or DALL-E, but for atoms instead of pixels. Additionally, the Affinity module, a special regression head, ‘stress tests’ the interaction prediction to provide a binding affinity score. Furthermore, inference-time steering sets physics-based guardrails on the simulation.
Simulating a Molecular “Glue”
Returning to our example of Rapamycin, we can see the output of Boltz-2 for simulating the ternary complex of the FRB domain of mTOR, Rapamycin, and FKBP12. This visualizes how Rapamycin ‘glues’ these two proteins together, affecting both of their functions. With the viewer, we can rotate and zoom in on the predicted interaction in three dimensions. Rapamycin doesn’t just find a pocket in the mTOR complex; it stabilizes the interaction between the protein and FKBP12.
Here, we have predicted the ternary structure with Boltz-2 using the CLI, with sequences from an early experimentally derived structure published in 1999.
After installing our Boltz-2 environment, we generate a YAML file for our experiment:
# rapamycin.yaml
version: 1
sequences:
- protein:
id: A # mTOR FRB Domain
sequence: VAILWHEMWHEGLEEASRLYFGERNVKGMFEVLEPLHAMMERGPQTLKETSFNQAYGRDLMEAQEWCRKYMKSGNVKDLTQAWDLYYHVFRRIS
- protein:
id: B # FKBP12
sequence: GVQVETISPGDGRTFPKRGQTCVVHYTGMLEDGKKFDSSRDRNKPFKFMLGKQEVIRGWEEGVAQMSVGQRAKLTISPDYAYGATGHPGIIPPHATLVFDVELLKLE
- ligand:
id: C # Rapamycin
smiles: 'C[C@@H]1CC[C@H]2C[C@@H](/C(=C/C=C/C=C\[C@H](C[C@H](C(=O)[C@@H]([C@@H](/C(=C/[C@H](C(=O)C[C@H](OC(=O)[C@@H]3CCCCN3C(=O)C(=O)[C@@]1(O2)O)[C@H](C)C[C@@H]4CC[C@H]([C@@H](C4)OC)O)C)/C)O)OC)C)C)/C)OC'
properties:
- affinity:
binder: C
After creating our YAML file, we use the Boltz CLI to generate the prediction:
boltz predict rapamycin.yaml --use_msa_server
The structure representation is interactive; you can rotate and zoom to explore the predicted structure and compare it to the published X-ray crystallography structure.
Benchmarking Affinity with Boltz-2
We know that the interaction of Rapamycin within this ternary structure is mediated by the FKBP12 protein, but can we verify this by simulating different combinations using Boltz-2? This represents a common use case where researchers probe cooperative binding in various contexts.
To scale this, we can use a batch process to programmatically call the Boltz CLI. Our workflow follows these steps:
- Generation: Programmatically use FASTA protein sequences to generate YAML configuration files for each experimental condition.
- Execution: Point the Boltz predictor to the input directory containing our generated YAMLs.
- Processing: Iterate through the results to compare the affinity binding scores across different protein combinations
After generating the different combinations of the YAML configuration files, we can programmatically call Boltz on the experiments and run the process.
import subprocess
def run_boltz(input_dir: str, output_dir: str, use_msa_server: bool = True):
cmd = ["boltz", "predict", input_dir, "--out_dir", output_dir]
if use_msa_server:
cmd.append("--use_msa_server")
subprocess.run(cmd, check=True)
run_boltz("inputs", "outputs", use_msa_server=True)
For this experiment, we performed five simulations for each combination to ensure statistical robustness. Why five? Structural predictions can vary slightly between runs but by averaging multiple seeds, we obtain a more reliable mean affinity score and can measure the variance.
It is important to note that a lower score is better, as the affinity prediction value represents the log10 of the half-maximal inhibitory concentration (IC50). A lower IC50 indicates a more potent binding interaction.
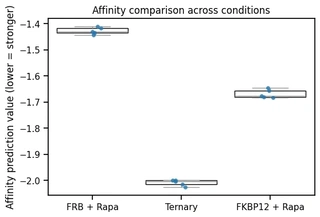
The simulation results clearly demonstrate the cooperative nature of the system. While Rapamycin shows affinity for both FKBP12 (-1.67) and the FRB domain (-1.43) individually, the ternary complex (-2.01) shows the highest predicted affinity. This significantly lower score confirms that the small molecule effectively stabilizes the interaction between the two proteins, acting as a potent molecular glue.
Accelerating Your Drug Discovery Pipeline
While models like Boltz-2 provide groundbreaking predictive power, there is often a significant gap between running a CLI tool on a research workstation and deploying a scalable, reliable drug discovery pipeline. A foundation model can provide the raw data, but the true value lies in how this data is integrated into a larger R&D workflow.
At Koehn AI we specialize in bridging the divide between cutting-edge science and robust software engineering. Whether you are looking to wrap these models in high-performance API endpoints, automate complex batch-processing workflows, or build intuitive 3D visualization tools for your researchers, we help you deploy AI that works in production.
Ready to accelerate your drug discovery pipeline? Contact us to discuss how we can help you build the infrastructure for the next generation of molecular science.